
Vasculitis UK is grateful to Dr Paul Brogan, Reader in Vasculitis and Honorary Consultant Paediatric Rheumatologist UCL Inst of Child Health and Great Ormond Street Hospital NHS Foundation Trust for preparing this article.
Introduction
The purpose of this article is to provide an overview of the different forms of vasculitis affecting children, summarising the most common clinical features, treatment, and recent advances (or lack of in certain diseases), and common diagnostic pitfalls for each condition.
IgA vasculitis (HSP)
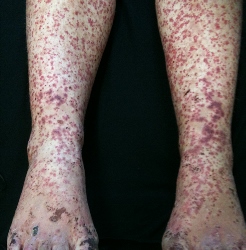
This is the commonest form of vasculitis in children, affecting between 10 and 20 per hundred thousand children every year. 50% of cases present before the age of five. Children usually present with rash (purpura, figure 1) predominantly affecting the lower limbs but occasionally much more widespread, tummy pain, arthritis and joint pains. The most severe complication is kidney involvement (glomerulonephritis), affecting about a quarter of patients, albeit usually only mildly. Less than 10% will develop more severe kidney involvement, sometimes requiring kidney biopsy and more intensive therapy and monitoring.
The vast majority of children do not require specialist investigations or treatment; simple medicines like paracetamol and or ibuprofen are helpful for relief from fevers, joint and abdominal pain. Those with severe abdominal pain, once the clinician is happy that there is not a severe complication called intussusception, may benefit from a short course of prednisolone.
Attempts to prevent the onset of renal involvement by giving prednisolone early in the disease have failed in clinical trials, although steroids and other immunosuppressants may have a role for those who do go on to develop severe kidney involvement. There are few clinical trials to guide treatment; corticosteroids undoubtedly help those with severe nephritis; the role of other immunosuppressants is not guided by evidence from clinical studies and therefore should be tailored to the individual patient under expert advice.
Pitfalls in the diagnosis and management of HSP
The trick is identifying which children have severe renal involvement, so that more aggressive treatment can be given earlier to secure better outcome for those rare cases. In general, children who present with evidence of kidney failure, lots of protein in the urine (sometimes with visible blood giving the urine a smoky red appearance), peripheral oedema and high blood pressure have the worst long-term kidney outcome; younger children tend to have a better outcome than older children and adults. Children with kidney involvement should be managed by a paediatric nephrologist.
Another common pitfall is to diagnose “atypical” HSP when in fact a more sinister form of vasculitis requiring much more aggressive treatment is required. The rash associated with polyarteritis nodosa, or ANCA vasculitis can be very similar to HSP. Clinicians should be alert to the possibility of more aggressive forms of vasculitis in children with unusually severe presentation of what initially looks like HSP.
Kawasaki disease (KD)
This is the second commonest vasculitic condition of childhood, affecting approximately 8 per 100,000 children under the age of five years, in the UK. The disease is much more common in the Japanese, and it is likely that genetic predisposition involving several genes leads to more reactive immune responses to infections, ultimately causing KD. The disease presents with prolonged fever, typically longer than five days, sometimes much longer, unusual rashes taking many forms (figure 2), red eyes, swollen lymph glands in the neck, red lips and tongue, swellings of the palms and soles with later peeling, and irritability. 25% of untreated children develop coronary artery aneurysms (figure 3); prompt treatment with intravenous immunoglobulin (IVIG) combined with high dose aspirin reduces this severe complication to approximately 4%; low dose aspirin in the convalescent phase may prevent thrombotic complications.
Recent clinical trials have demonstrated that those with more severe disease have even higher risk of coronary artery aneurysms, and benefit from the addition of corticosteroids to IVIG to prevent coronary aneurysms. A new UK guideline to help clinicians identify which patients require corticosteroids to be added to IVIG is soon to be published.
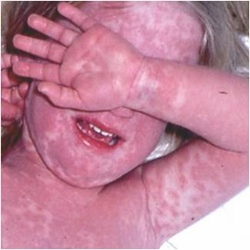
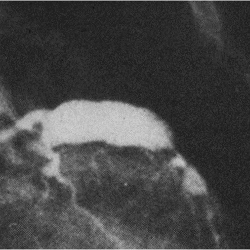
Pitfalls in the diagnosis and management of KD
In the past it was stated that “if you found evidence of infection, then you couldn’t diagnose KD”. We now know that infectious triggers are relatively common for KD; clinicians should not be put off the diagnosis if the clinical features fit and there is a lack of response to antibiotics, even if an apparent infection is discovered at the disease onset. If there is diagnostic uncertainty, children can receive antibiotics and IVIG to prevent severe coronary artery injury from missed KD.
Another important point to emphasise is that the clinical features may present sequentially; clinicians should be alert to this, and enquire specifically about retrospective symptoms or signs that may fit with KD but have resolved by the time of clinical review. Lastly, another misconception is that “treatment doesn’t work 10 days after the onset of the illness”; this is incorrect, and clinicians should treat after 10 days if there are features of ongoing inflammation indicated by blood tests, and/or other inflammatory clinical features.
Polyarteritis nodosa (PAN)
This is a rare but serious form of vasculitis, involving predominantly medium and small-sized arteries. The clinical features may evolve gradually with non-specific features such as fever, rashes and joint pains. The disease can affect any organ in the body, including the gut, brain and nerves, and kidneys. PAN is a great mimic of many diseases affecting children; chronic infection, malignancy such as leukaemia, immunodeficiency, and other forms of vasculitis such as HSP need to be excluded. Specialist tests such as skin/ other tissue biopsy, nerve conduction studies, and specialist imaging including various forms of angiography are important to secure the diagnosis.
Some children develop a predominant cutaneous form of the disease, requiring less aggressive approaches to treatment. Children with severe forms of the illness should, however, be treated aggressively with high doses of corticosteroids and immunosuppressants such as cyclophosphamide to induce remission. After 3 to 6 months, the treatment can be changed to lower doses of corticosteroids and milder forms of immunosuppression such as azathioprine to maintain remission for a further 1 to 2 years, depending on the clinical course.
Although relapses can occur, unlike ANCA vasculitis there is a very good chance of curing PAN using this approach. The first ever clinical trial for paediatric PAN, comparing mycophenolate mofetil vs cyclophosphamide (the MYPAN trial), is currently being set up, with planned patient recruitment to begin in July 2014.
Pitfalls in the diagnosis and management of PAN
The commonest pitfall is to confuse severe systemic PAN with HSP, or the more benign cutaneous form of PAN. This leads to therapeutic delay and worse outcomes. Another misconception is that “non-invasive” forms of angiography such as CT angiography, are equivalent to catheter angiography, were x-ray dye is actually inserted into the arteries (usually under general anaesthetic) to obtain a detailed picture of vascular tree; the latter is much more sensitive for the detection of PAN than the former (Figure 4). Clinicians therefore should not be falsely reassured by a normal CT angiogram, and should seek expert advice to consider formal catheter angiography from centres used to doing this test in children if there is diagnostic uncertainty.
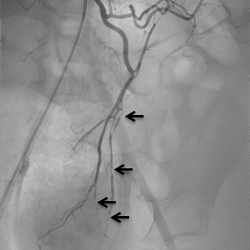
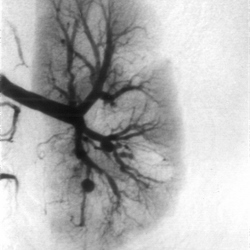
More subtle changes, including calibre variation and beading of medium-sized mesenteric arteries are 4A: selective renal digital subtraction arteriography in an eight-year-old girl with polyarteritis nodosa. Large aneurysms, small aneurysms, renal, parenchymal perfusion defects, and calibre variation of intrarenal arteries demonstrated. Perfusion defects in the renal cortex are also present.
4B: selective mesenteric digital subtraction arteriography in a six-year-old boy with partially treated polyarteritis nodosanstrated (arrowed) and these are unlikely to have been detected using non-invasive angiography such as CT angiography or MR angiography.
ANCA associated vasculitis (AAV)
Granulomatosis with Polyangiitis (GPA, formerly Wegener’s granulomatosis) occurs in children much more commonly than microscopic polyangiitis (MPA), although both are rare forms of vasculitis, probably affecting one per million children per year. Approximately 2 new children a year are seen at Great Ormond Street Hospital.
The clinical features of GPA in children are virtually identical to those affecting adults, with ear nose and throat inflammation, airway involvement, lung involvement, and renal vasculitis, amongst other symtoms. Data from clinical trials in adults informs the current approaches to treatment in children: induction of remission with high-dose corticosteroids combined with cyclophosphamide, rituximab, or mycophenolate mofetil (MMF); followed by maintenance of remission with much lower doses of corticosteroids combined with azathioprine, MMF, or other immunosuppressant.
The MYCYC trial was the first EUVAS trial to include children, and compared MMF vs cyclophosphamide for the induction of remission; the results will be published later this year. The PEPRS trial is currently examining the use of rituximab as an induction of remission agent in children with AAV, and recruitment began in 2013.
Pitfalls in the diagnosis and management of AAV in children
The main pitfall is delay in diagnosis leading to organ injury and in some cases even death in the pre-diagnostic phase of the illness. Ear, nose, and throat symptoms are very common in the paediatric population, and GPA may not be considered in younger children as a potential cause. More widespread availability of ANCA testing and increased clinician awareness will reduce diagnostic delay, leading to earlier treatment and better outcomes.
Takayasu arteritis (TA)
This is the only recognised large vessel vasculitis affecting children. The disease is very rare, probably affecting less than one in 1 million children per year. The disease is more common in the Japanese population, and previous suggested links to tuberculosis or other infectious trigger have not been confirmed. The disease presents very non-specifically with symptoms such as fevers, headache, fatigue, aches and pains. Hypertension may be the first indication of a problem in children.
It is useful to consider the illness in two phases: the initial inflammatory vasculitic phase, followed by a later fibrotic stenotic phase of the illness, where large arteries become progressively narrowed leading to symptoms due to lack of blood supply to organs and limbs. Due to the non-specific nature and rarity of the condition, children are often well into the stenotic phase of the illness by the time the diagnosis is made, sometimes many years after the disease onset.
Angiography, and other forms of imaging are key to securing the diagnosis. Treatment usually involves corticosteroids combined with another immunosuppressant (such as cyclophosphamide, methotrexate, MMF etc.) to induce remission; followed by tapering corticosteroids usually combined with methotrexate or azathioprine to maintain remission. Anti-TNF and anti IL-6 treatments are beginning to be used for resistant paediatric cases, but there has never been a clinical trial to guide the management of children or adults with this rare form of vasculitis.
Pitfalls in the diagnosis and management of TA in children
Clinicians should be alert to the possibility of TA in patients with unexplained inflammation and cardiovascular symptoms such as unexplained cardiac failure, hypertension, or other symptoms of vascular insufficiency affecting the limbs or organs. The disease can even present in infancy; the heart may be primarily affected in children with heart valve insufficiency, myocarditis, or even ventricular aneurysms. Another major diagnostic challenge even when the disease is considered, is to differentiate TA from the many non-inflammatory forms of vasculopathy (sometimes with genetic cause) that can affect the paediatric population, the commonest being the entity fibromuscular dysplasia.
Once the diagnosis of TA is secured, another challenge is how to monitor disease activity; blood tests such as ESR and CRP may not be reliable; magnetic resonance scans of arteries looking for features of large vessel vasculitis are important in this context, as are other approaches to imaging such as PET scans, but require specialist interpretation.
Unclassified forms of vasculitis
There are many other forms of vasculitis affecting children that don’t fit into any single entity. It’s essential that clinicians do not delay therapy if severe vasculitic manifestations are unfolding, even if they don’t fit into any particular diagnosis. The sage advice of “treat now and ask questions later” may apply here.
Paediatric vasculitis guidelines
Summary
The last 10 years or so has seen important developments and advances in the field of paediatric vasculitis: notably an increase in the number of clinical trials, particularly in relation to Kawasaki disease, but also for more rare forms of paediatric vasculitis including the first trials in AAV and PAN for children; development and validation of classification criteria for paediatric vasculitic diseases; and development and validation of a tool to assess clinical disease activity in children (paediatric vasculitis activity score, or “PVAS”). We still have much more to do however; in particular diagnostic delay is still contributing to poor outcomes. There therefore needs to be increased awareness particularly of the rarer and more serious forms of vasculitis that can affect children.